Saira Qurban, Hao-Wen Jiang, and Peng-Fei Xu*
https://doi.org/10.1002/chem.202501961
abstract
One of the most difficult issues facing humanity today is thetreatment of cancer. A binary cancer treatment called boronneutron capture therapy (BNCT) works especially well for high-grade gliomas and metastatic brain malignancies. Due to theirpreferential absorption by developing tumor cells, boronatedamino acids have drawn a lot of attention among the severalboron-containing compounds utilized as BNCT agents. In the first section of this review, the fundamental ideas of BNCT arebriefly presented. In the remaining sections, we present a thor-ough analysis and synthesis of several boronated amino acidsusing distinct techniques. This article summarizes the thera-peutic prospects of boron delivery agents and offers a criticalanalysis of them from the perspectives of nuclear medicinedoctors and medicinal chemists.
Huan-Huan Zhao, Xu-Gang Zhang, Hao-Wen Jiang, Yi Zhao, Xiu-Qin Hu,*and Peng-Fei Xu*
https://doi.org/10.1021/acs.orglett.5c03480
ABSTRACT:
Inthisstudy,wedevelopedavisiblelight-mediatedC(sp3)−Haminationstrategyenabledbyligand-to-metalcharge transfer(LMCT)andironcatalysis.TheinexpensiveFe(NO3)3·9H2Oaffordsaphotoactiveironcomplexuponcoordinationwith N-heteroarylsubstrates,andthecomplexundergoesLMCT-inducedβ-homolysistoproduceahigh-valentFe(IV)�Ospecies.The Fe(IV)�Ospeciespromotesalkyl radical formation,enablingefficientC(sp3)−Nbondconstruction.Thereactionoperatesunder mild conditionswithbroad functional group tolerance and excellent scalability, providing a practical platformforC(sp3)−H amination.

Xiang-Chuang Tan, Lei Yan, Hao-Ni Qin and Peng-Fei Xu*
https://doi.org/10.1039/D5GC03425F
Abstract
Herein, we report the development of a carbon-bound radical cyanating reagent, 2-methyl-2-(pyridin-2 yl)malononitrile (MPYMN), which enables metal-free photocatalytic cyanation with high reactivity and site selectivity. MPYMN is a bench-stable, readily accessible solid that does not release cyanide during reac tions nor does it require metals or harsh conditions. It exhibits superior performance compared to com mercially available cyanating agents and other carbon-bound cyanide sources. Importantly, the steric effect of the quaternary carbon in MPYMN facilitates reagent-controlled site-selective cyanation. The syn thetic utility of this reagent is demonstrated by its compatibility with both photoredox and hydrogen atom transfer (HAT) processes, including efficient late-stage functionalization of complex molecules relevant to medicinal chemistry.

Yi-Xiang
Geng, Teng-Fei
Xiao, Dong Xie, Ming-MingLi, Pan-Pan
Zhou, Guo-Qiang Xu*, Peng-Fei Xu*
https://doi.org/10.1038/s41467-025-64399-7
Abstract
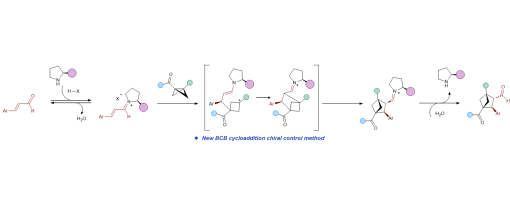
Bicyclo[2.1.1]hexanes (BCHs), three-dimensional benzene bioisosteres char acterized by high sp3-carbon content, hold great promise for diverse appli cations in medicinal chemistry. Although significantadvanceshavebeenmade in the synthesis of racemic BCHs, highly enantioselective approaches remain comparatively rare. Here we report a mild, secondary amine–catalyzed asymmetric [2π+2σ] cycloaddition of bicyclo[1.1.0]butanes (BCBs) with α,β-unsaturated aldehydes, whichovercomeskeylimitationsofexistingmetal catalyzed and photochemical methods. The protocol operates under ambient air and tolerates a wide range of BCB and aldehyde substrates bearing diverse functional groups, affording BCH scaffolds in yields of up to 84% under Supramolecular Iminium Catalysis with excellent enantioselectivity (up to 99% ee) and high diastereoselectivity (>20:1 dr). The mild conditions and opera tional simplicity underscore the potential of this transformation for stereo selective manufacturing of BCHs at scale. Mechanistic experiments and DFT studies support an acid-promoted dual activation of both substrates, followed by an enamine–iminium tandem catalytic process that delivers the enan tioenriched products.
Zhi Lei, Hao Tang, Heng Zhang, Yong-Chun Luo*, Peng-Fei Xu*
http://10.1016/j.psep.2025.107561
ABSTRACT
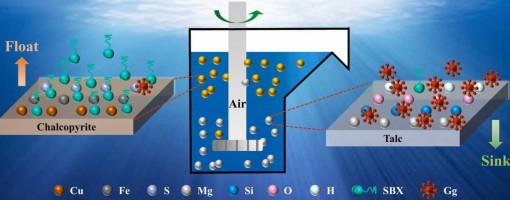
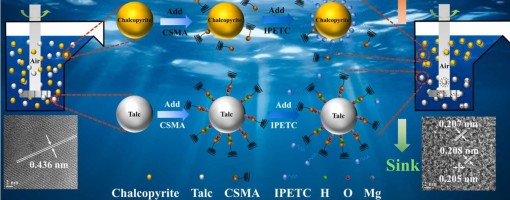
The intergrowth phenomenon of chalcopyrite and talc makes the identification of an eco-friendly and efficient depressant crucial for achieving effective separation. In this study, we innovatively employed Ghatti Gum as a talc depressant to improve the flotation separation. The micro-flotation test results demonstrate that, compared to conventional depressants, Ghatti Gum exhibits a stronger depressing effect on talc while maintaining excellent selectivity toward chalcopyrite. Atomic force microscopy (AFM), scanning electron microscopy with energy- dispersive X-ray spectroscopy (SEM-EDS), and transmission electron microscopy (TEM) images confirmed that Ghatti Gum adsorbed selectively on the surface of talc, which significantly altered its surface roughness and elemental composition. In contrast, Ghatti Gum exerts a weaker influence on chalcopyrite. Further analysis through Zeta potential, Fourier transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS), and density functional theory (DFT) calculations revealed that Ghatti Gum adsorbs onto the talc surface through hydrogen bonds and hydrophobic interactions. Meanwhile, Ghatti Gum has less influence on the elemental composition and floatability of the chalcopyrite surface. In summary, Ghatti Gum emerges as a promising eco- friendly depressant for the flotation separation of chalcopyrite and talc. This discovery not only expands the applications of Ghatti Gum but also provides novel insights for the efficient separation of chalcopyrite from talc.
Zhi Lei, Heng Zhang, Hao Tang, Yong-Chun Luo*, Peng-Fei Xu*
http://10.1016/j.jece.2025.117093
Abstract
The flotation separation of chalcopyrite from talc presents a significant challenge due to its analogous surface characteristics. This study investigated the selective depression of talc in the presence of chalcopyrite using a novel modified chitosan (CSMA), achieving effective mineral separation. CSMA possesses a porous architecture compared to unmodified chitosan (CS), which facilitates enhanced interaction with mineral surfaces. Flotation tests revealed that CSMA significantly inhibited talc flotation while exerting minimal impact on chalcopyrite recovery. Morphological and elemental changes were analyzed using inductively coupled plasma optical emission spectroscopy (ICP-OES), atomic force microscopy (AFM), scanning electron microscopy with energy-dispersive X-ray spectroscopy (SEM-EDS), and transmission electron microscopy (TEM). These analytical results confirm that CSMA adsorbs onto the talc surface. In addition, zeta potential measurements, Fourier transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS), and density functional theory (DFT) calculations collectively indicate that CSMA exhibits enhanced reactivity, primarily through hydrogen bonding and hydrophobic interactions with the talc surface. This investigation elucidates the mechanism by which carboxymethyl chitosan acts as an effective depressant in the flotation separation of copper sulfide minerals, thereby establishing a foundation for the environmentally friendly recovery of these minerals.
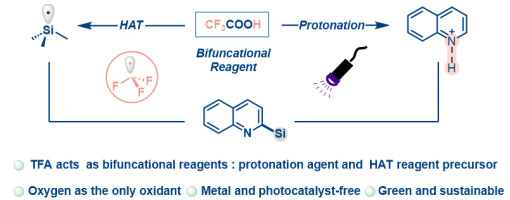
Rui Zhang, ‡ Hao-Wen Jiang, ‡ Xiu-Qin Hu, and Peng-Fei Xu*
https://doi.org/10.1021/acs.orglett.5c03441
ABSTRACT:
A photoinduced Minisci reaction involving Si−H bond activation via a CF 3 radical generated from trifluoroacetic acid as a hydrogen atom transfer reagent has been achieved for the f irst time. This approach enables the synthesis of a diverse array of silylated electron-deficient heteroarenes in moderate to high yields. Mechanistic studies were conducted to provide supporting evidence for the reaction process. The reaction features a metal free protocol, operational simplicity, and compatibility with mild reaction conditions.
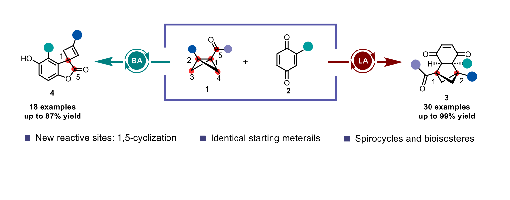
Ming-Ming Li, Teng-Fei Xiao, Yi-Xiang Geng, Guo-Qiang Xu,* and Peng-Fei Xu*
doi.org/10.1021/acs.orglett.5c02160
ABSTRACT:
Bicyclo[1.1.0]butanes (BCBs), which possess multiple switchable reactive sites, serve as the most direct modular scaffolds for constructing benzene ring bioisosteres. Herein, we describe the precise modulation of BCB reactive sites through Lewis/ Brønsted acid switching to enable the synthesis of challenging spirocycles and bridged frameworks. The divergent reaction outcomes, arising from the precise control of catalysts over the switchable reactive sites of BCBs, are achieved without any substrate modification
Lei Yan, Hui-Qing Yang, Wan-Lei Yu, Xu-Gang Zhang, and Peng-Fei Xu*
https://doi.org/10.1021/jacs.5c03154
ABSTRACT:
Radical asymmetric reactions represent a crucial strategy in asymmetric synthesis, which is characterized by their high reaction efficiency and unique reactivity profiles. Despite significant progress in radical-based asymmetric transformations, the formation of C-N bonds using nonredox metal complexes via the inner-sphere stereocontrol mechanism remains a formidable challenge in the development of novel asymmetric catalytic strategies. This study introduces an innovative and highly efficient asymmetric photochemical bifunctional catalysis that utilizes a combination of magnesium salts and chiral PyBOX-type (pyridinebisoxazoline) C2-symmetric ligands under visible light irradiation. This approach enables the selective α-amidation of β-keto esters via an N-centered radical mechanism, facilitating the synthesis of substituted β-keto amino acid derivatives with a fully substituted stereocenter. The reaction proceeds in good yields (up to 79%) and excellent enantioselectivity (up to 94%). The catalysis proceeds through the in situ formation of prochiral quaternary charge-transfer complexes, which promote the Lewis acid-supported generation of radicals, thereby mediating the subsequent enantioconvergent radical-radical cross-coupling. Notably, the β-keto ester serves a trifunctional role as a sensitizer, reductant, and radical precursor, while the N-protected iminopyridinium ylide functions as both the oxidant and N-centered radical precursor. Experimental and computational mechanistic studies corroborate the enantioconvergent radical-radical cross-coupling process.

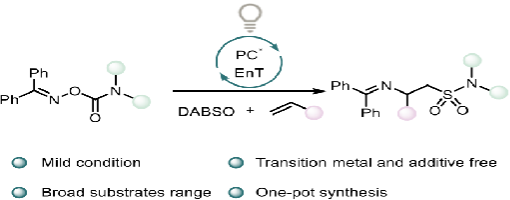
Ai-Lian Wang, Huan-Huan Zhao, Hao-Wen Jiang, and Peng-Fei Xu*
https://doi.org/10.1021/acs.orglett.5c01128
In this study, we have devised a strategy that employs oxime carbamate as a bifunctional diamination reagent in combination with SO2 to realize imino-sulfamoylation of alkenes. This protocol is characterized by its mild conditions, operational simplicity, and metal-free nature, while demonstrating broad functional group tolerance for alkenes. Furthermore, the application of this method provides an accessible route to a diverse range of β-amino sulfonamide derivatives.